液相色譜常見問題與解答
1何謂反相柱、正相柱?
答:“反相”和“正相”的概念是液相色譜法早期提出的概念,當時鍵合相色譜柱尚未出現,固定相被涂覆在載體表面,極易流失,為此科學家對流動相使用給出了合理的建議:流動相極性與固定液極性應具有較大差別,以減少固定液流失。固定相極性弱于流動相時的液相色譜法被稱為反相色譜法,固定相極性強于流動相時的液相色譜法被稱為正相色譜法。盡管目前鍵合相色譜柱已成為主流,但這一概念在色譜方法開發、預測出峰順序等方面具有重要意義。
由上面的介紹可知具體的色譜方法、色譜柱屬于正相還是反相不僅取決于固定相極性,同時還取決于流動相極性。C18(硅膠鍵合十八烷基硅烷)、C8(硅膠鍵合辛基硅烷)、PH(硅膠鍵合苯基硅烷)等色譜柱,由于固定相極性極低,比目前已知的任何流動相的極性都要低,因而是標準的反相柱;Silica(硅膠)、NH2(硅膠鍵合氨丙基硅烷)具有較高的極性,主要用于分離帶有極性基團的化合物,所用流動相的極性通常低于這些固定相,因而是標準的正相柱。CN(硅膠鍵合腈丙基)的極性適中,當流動相極性超過CN時,它屬于反相柱,反之則是正相柱。
2色譜柱規格對分析結果會產生何種影響?
答:色譜柱內徑決定載樣量,載樣量與內徑的平方成正比;色譜柱長度與塔板數成正比,與柱壓成正比;粒徑影響渦流擴散相,粒徑越小渦流擴散相越小,柱效越高,粒徑與柱效近似成反比;粒徑越小,壓力也越大,壓力與粒徑的平方成反比。填料孔徑對分析對象的分子量有限制,當孔徑為分析物尺寸的5倍以上時,分析物才能順利通過孔隙,孔徑處于60~120Å的色譜柱適用于相對分子量小于10000的分析物,孔徑為300Å的色譜柱可以滿足分子量處于10000以上的大分子化合物分析。
3液相色譜分析中如何才能提高分離度?
答:下式為分離度計算公式
N:柱效(Efficiency)反映色譜柱性能,柱效越高,分離度越好。在其他條件恒定的情況下,塔板數增加一倍,分離度僅提高40%。操作中,可通過下面兩種方式增加塔板數進而提高分離度:其一,使用長柱或雙柱串聯,但也會使分離時間大大延長;其二,使用細粒徑填料的色譜柱,但這需要耐更高壓力的液相色譜系統。相比之下后者更為可取。
α:選擇性(selectivity)是指色譜柱-流動相體系分離兩個化合物的能力。選擇性主要與固定相、流動相組成以及柱溫等因素有關,與保留值也密切相關,其中固定相和流動相組成影響較大。以zui常見的反相模式為例,反相柱(包括C18、C8、PH等)是以分配作用對化合物進行保留的,不同化合物的分離是基于它們在鍵合相與流動相中分配系數的差異,如果兩種化合物的水溶性、在烷烴-水體系的分配系數等方面存在明顯差異,那么這些化合物通常是能夠利用反相柱達到分離;PH柱對具有苯環的化合物具有特殊保留。正相模式下,硅膠柱、胺基柱、氰基柱與帶有極性基團的化合物之間存在極性相互作用,對化合物的基團具有選擇性,常常用于結構類似物、異構體化合物的分離。流動相方面,降低流動相的洗脫強度通常可以增大分離度;而有機溶劑類型也會影響分離,比如反相條件下,乙腈和甲醇的選擇性就存在很大差異,這種差異需要在實踐中摸索,但無論如何,多種溶劑類型帶給我們更多的實現分離的可能。
k:隨著容量因子k的增大,分離度也隨之增加,這種影響在k值較低時非常明顯,當k值大于10時,k值增加對分離度的影響就不再顯著,這就告誡無原則地提高k值以增大分離度是沒有意義的。增加鍵合相密度能夠提高k值;另外改變鍵合基團類型也能改變k值,比如在反相色譜中,隨著鍵合相碳鏈長度的增加,k值逐漸增大。
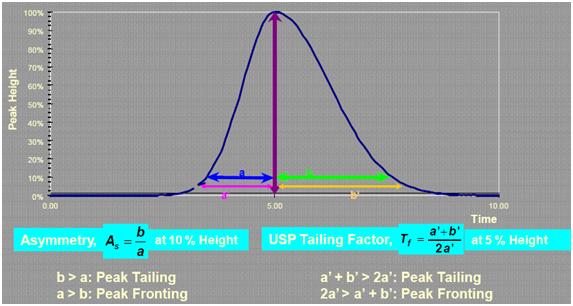
4色譜峰的峰形是怎樣衡量的?有何規定?
理論上講,色譜峰應符合高斯曲線分布,然而實際上任何色譜峰都對高斯曲線分布存在一定的偏離,亦即不對稱。峰拖尾可以用不對稱因子(As)或USP拖尾因子(Tf)來衡量,顯然不對稱因子的說法更準確,因為色譜峰存在前延、對稱、拖尾三種形態。一般來說,制藥行業以USP拖尾因子作為評測標準,而其他行業則多采用As來測定峰形。
對于藥物分析,通常有明確的規定,Tf應處于某一范圍之間,比如我國藥典規定某些藥物的拖尾因子應處于0.95~1.05之間。其他行業尚無較為明確的規定。
5什么是梯度洗脫?什么情況下使用梯度洗脫?
答:為了改善分析結果,某些操作需要連續改變流動相中各溶劑組分的比例以連續改變流動相的極性,使每個分析組分都有合適的容量因子k,并使樣品種的所有組分可在zui短時間內實現*分離,這種洗脫方式稱為梯度洗脫。
梯度洗脫可在下列情形中發揮重要作用:
A在等度下具有較寬k值的多種樣品分析。
B大分子樣品分析。
C樣品含有強保留的干擾物,在目標化合物出峰后設置梯度洗脫,將干擾物洗脫出來,以免其影響下一次數據采集。
D分析方法建立時,不知道其洗脫情況,使用梯度洗脫,找出其較優的洗脫條件。
6何謂封端?封端的意義是什么?
硅膠表面的硅羥基(Si-OH)密度為8μmol/m2,由于空間位阻的存在,硅烷化鍵合反應zui多只能覆蓋50%的硅羥基,超過一半的硅羥基是活性硅羥基。這些硅羥基與化合物的極性基團存在極性相互作用和離子交換作用,為化合物的保留增加了不被期望的作用力,往往會影響峰形。用短鏈氯硅烷(如*基氯硅烷)鍵合活性的硅羥基,可以減小甚至消除這種影響,這種操作被稱為封端(Endcap)。
封端處理的意義:
抑制了特異性吸附,提高了色譜峰的對稱性,并改善了分離效果;
在一定程度上掩蔽了硅膠表面,使其對堿性環境的耐受性增強;
通過空間位阻掩蓋了鍵合反應形成的Si-O-Si鍵,使其對酸性環境的耐受性增強;
可能會影響對極性樣品的選擇性。
7柱床塌陷和鍵合相塌陷
柱床塌陷是指色譜柱使用一段時間后色譜柱入口處的柱床產生可見的空隙。該空隙的存在增大了死體積,會導致色譜柱柱效下降。造成柱床塌陷的原因如下:其一,色譜柱填裝時的壓力過低,填裝不緊密,在高壓下使用一段時間,開始出現空隙;其二,操作壓力超出色譜柱填料的耐壓值,導致填料顆粒破碎產生空隙;其三,流動相溶解填料導致空隙出現。鍵合相塌陷是指由于流動相極性與鍵合相極性相差太大,鍵合相無法在流動相中充分伸展而倒伏、纏結在一起,比如普通C18在純水相
條件下。相塌陷會導致色譜柱對化合物的保留不足。
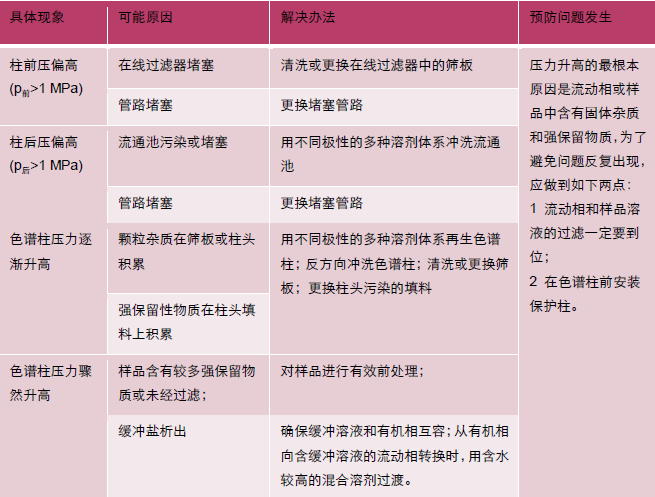
8系統壓力升高的原因及排除
使用者通過觀察儀器的系統監測掌握系統的壓力,如果發現壓力偏高,不要立即認為“柱壓高了”,因為系統壓力通常由柱前壓力、色譜柱壓力和流通池壓力加合而成。此時正確的做法是:在操作條件下,測定系統壓力,得到p總;卸下色譜柱,在相同條件下,測定柱前壓力,得到p前;用兩通將泵流出管路與流通池連接,在操作條件下,測定壓力,得到p(前+后)。通過計算找出壓力高是來自柱前、色譜柱還是柱后。
以上情形假設沒有安裝保護柱,如果安裝了保護柱,壓力升高時應先測試保護柱的壓力,以便確認問題是否來自保護柱。
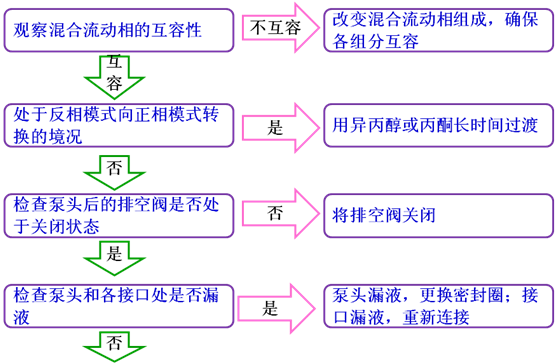
9系統壓力不穩定
通常情況下,泵壓的變動值超過了0.5Mpa,系統壓力就屬于不穩定的范疇。導致系統壓力不穩定的zui直接原因為流動相流量和組成輸出的不穩定,而導致流動相輸出不穩定的原因通常包括溶劑互容性較差、系統漏液、系統存在氣泡以及輸液系統部件工作失效等等。下面將介紹排除“系統壓力不穩”的方法。
流動相輸出不穩定還會導致基線不穩定以及保留時間變化等現象,所以基線不穩定或保留時間不重現時,先看一下柱壓是否穩定,如果不穩定,三個問題可以合并處理。
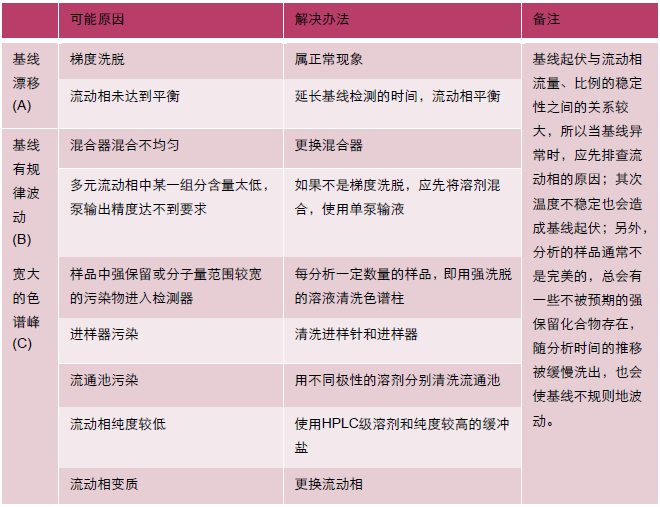
10基線不穩定
基線不穩定是指色譜曲線在較長時間范圍內對響應值的0刻度較大的偏離,通常包括基線漂移(單方向)、基線起伏(規律的上下起伏)、區域寬度極大的色譜峰三種情況
| 問題 | 現象 |
| A 基線漂移 |  |
| B 基線波動 | 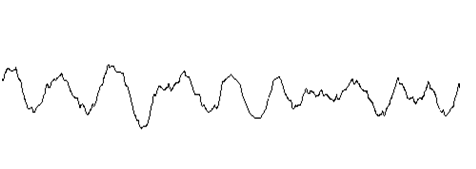 |
| C 寬大色譜峰 | |
如果基線不穩定同時伴隨壓力波動,請按照“壓力不穩定”進行排查。如果壓力穩定,請按下表中的方法進行排查。
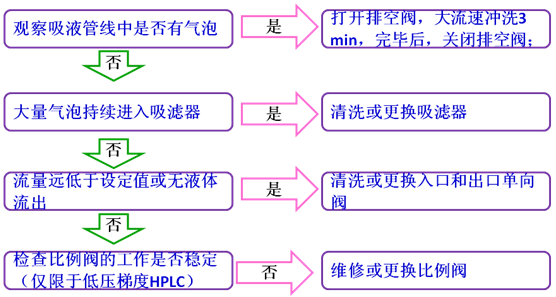
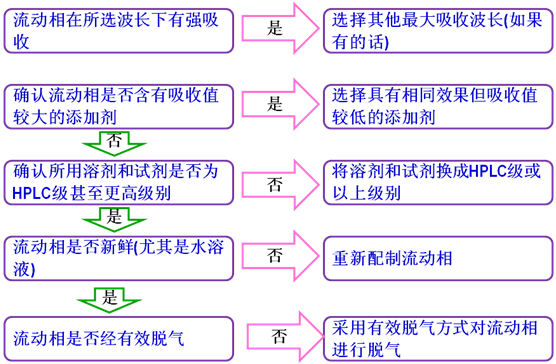
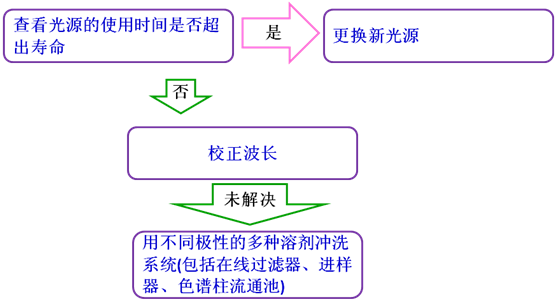
11噪聲升高
噪聲(baselinenoise):由檢測器輸出與被測樣品組分無關的無規則波動信號,在特定靈敏度下用響應單位表示。可分為高頻噪聲和短周期噪聲兩種。對于光譜型檢測器,噪聲的正常響應值處于0.000005V~0.00002V之間(參考Shimadzu的HPLC儀器),高于此值,即可認為噪聲偏高。
噪聲升高時,先按下列步驟排查流動相問題
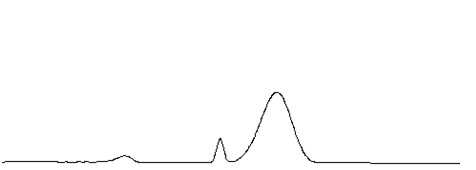
12柱性能下降
柱性能下降的一個重要現象就是在保留時間基本不發生變化的情況下,色譜峰的區域寬度明顯增大(如下圖)。
當遇到柱效下降,請按下表進行排查和解決
13保留時間重現性差
當保留時間的偏差超過2%,可以認為保留時間的重現性差。對于某一個特定的分析,保留時間主要受流動相組成、流速、溫度以及固定相影響,當保留時間發生明顯變化時,應從這幾方面入手。
A由流動相造成的保留時間不穩定
B 由固定相造成的保留時間不穩定
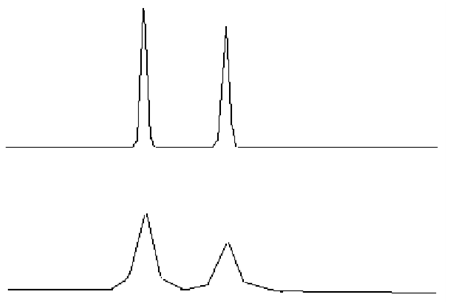
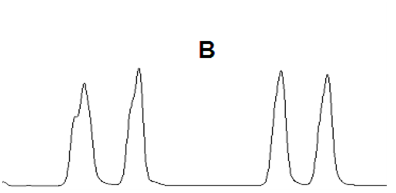
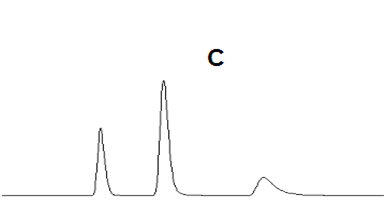
14峰形不理想
理想的色譜峰遵循正態分布,應該符合高斯曲線,是而對稱的峰形(如圖A);然而由于化合物性質的特殊性、色譜柱故障、操作條件等問題存在,峰形可能會不對稱,前延(B)或拖尾(C)。
峰形前延(B)
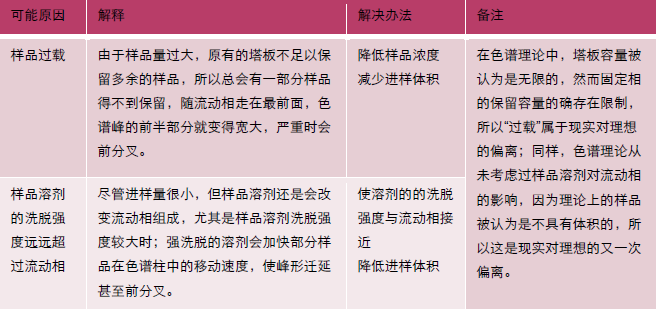
中藥成分分析項目中,提取溶劑通常為乙醇、甲醇、乙酸乙酯等洗脫強度較強的溶劑,此時易發生“樣品溶劑的洗脫強度遠遠超過流動相”的情況,為避免前延峰出現,可以用通過“流動相稀釋提取液”或降低“進樣體積”的方式。
峰形拖尾(C)
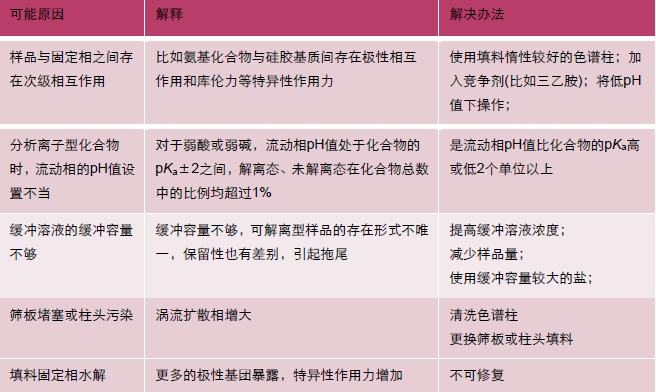
15鬼峰
鬼峰(Ghostpeak):是對未知來源的色譜峰的統稱。

16氣泡峰
氣泡峰是指攜帶氣泡的流動相通過檢測器時得到的響應值;這里的氣泡可能是流動相中釋放的空氣,也可能是有機相遇水放熱而會釋放的有機溶劑分子;氣泡峰往往具有較高的響應值,區域寬度僅為數秒,出峰時間和相應值比較有規律(如下圖)。通常只有光譜型檢測器才會出氣泡峰。

解決辦法:
脫氣
使用多元梯度時,對每一種組分進行脫氣,排除它們溶解的空氣;
對于預先混合的溶劑,混合后脫氣,排除溶解的空氣和因放熱而揮發出的有機溶劑分子;
前面的脫氣方式對于預先混合好的混合溶劑,基本可以達到消除氣泡峰的目的;但對于多種純溶劑在儀器中混合的情況,混合后溶劑體系放熱,存在潛在的氣泡,當流動相流出色譜柱時,壓強驟減,潛在的氣泡溢出,被檢測器檢測到。為了防止這一問題出現,應在檢測器出口連接一段內徑較細的管路,使流動相通過檢測器時,環境壓強較高,氣泡難以溢出,可以避免氣泡峰出現。

.bmp)

")